Blood Unit 11 & 12
Platelets, Coagulation &
Bleeding Disorders
Platelet
• Platelets are produced in blood cell formation (thrombopoiesis) in bone marrow• megakaryoblast > pro-megakaryocyte > immature megakaryocyte > megakaryocyte > Platelet
• Platelets or thrombocytes are small in size
• irregularly shaped
• non-nucleated
• 2-3 µm in diameter.
• lifespan of circulating platelets is 8 to 12 days.
• Normal Value - 1.5 to 4 lakhs/uL
• platelet production is regulated by thrombopoietin (hormone which produced by the liver and kidneys)
• Old platelets are destroyed by phagocytosis in the spleen and by Kupffer cells in the liver


• The platelet structure has 3 zones:
- Peripheral
- Structural
- Organelle
• Structural zone
- Consists of the cytoskeleton
- The cytoskeleton forms the support for the maintenance of the platelet's discoid shape
- Regulate contractile system that allows, upon activation, shape change, pseudopod extension, internal contraction, and release of granular constituents.


• Organelle zone
- consists of the granules and cellular components
- These organelles serve in the metabolic processes of the platelet and store enzymes.
- dense granules contain non-metabolic adenosine triphosphate (ATP) and adenosine diphosphate (ADP), serotonin, and calcium
- alpha granules contain adhesive proteins such as fibrinogen, fibronectin, von Willebrand factor (VWF), thrombospondin.
- alpha granules also contain growth-promoting substances such as platelet-derived growth factor (PDGF), platelet factor 4, and transforming growth factor.
- Coagulation factors including factor V, high molecular weight, factor XI, and plasminogen activator inhibitor-1 are also present in the alpha granule.
• Membrane / peripheral zone
- Consist of typical phospholipid bilayer membrane
- Embedded in this structure are different kind of glycoprotein.


General function of platelet
• The function of platelets is the maintenance of hemostasis.• Platelets helps in blood clotting.
• Wound repair
• Platelets secrete platelet-derived growth factor (PDGF).
• Granule secretion.
• Adhesion and aggregation.
• Pro-coagulation.
• Cytokine signalling.
• Phagocytosis.
• Transport of enzyme and proteins critical to clotting.
• Formation of a platelet plug to slow blood loss.
• Contraction of a clot after it has formed, which then reduces the size of the vessel break.




Platelet Adhesion
• Injured vessel get attached with vWF on the endothelium cells which attracts platelets to get attached to vWF
Platelet Activation
• Adhered platelets gets activated and changes there shape into pseudopodia, discharges granules to get more platelets attracted.Platelet Aggregation
• Activation of phospholipase C into Phospholipase A2.• It causes more platelet aggregation and formation of temporary haemostatic plug.
• This loose plug later gets into secondary Haemostatic Plug by fibrin.
• Fibrin is activate via activation of different clotting factors.








Variations in Count
• Thrombocytosis- Increase in Platelet Count• Causes- Administration of Epinephrine
Trauma
Removal of Spleen
• Thrombocytopenia- Decrease in Platelet Count
• Causes- Bone Marrow Depression,
Hypersplenism, Viral Infections, Leading Purpura
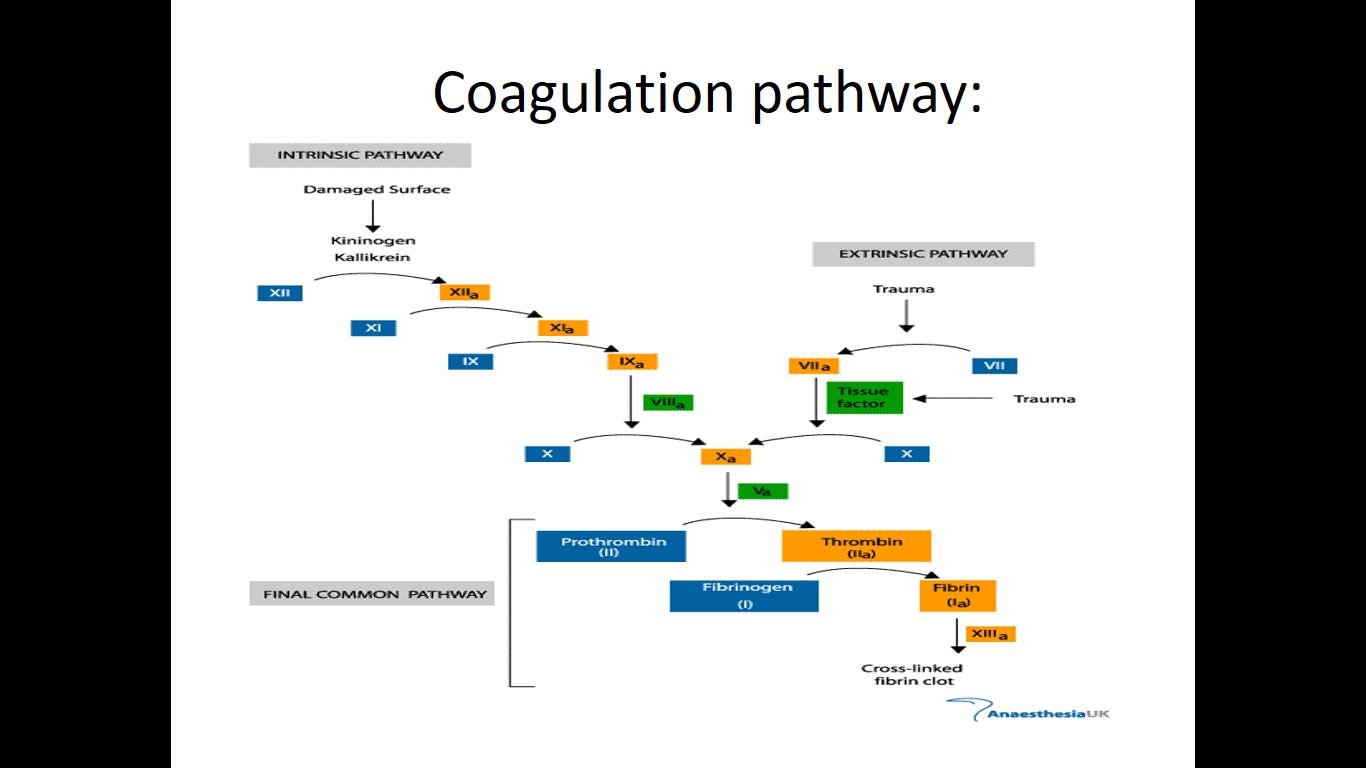
Types of Bleeding disorder:
• Divided into:- Coagulation disorder
- Platelets disorder
• Coagulation disorder include:
- Hemophilia
- Von Willebrand disease • Platelet disorder include:
- Deficiency Of Vitamin K.
- Thrombotic Thrombocytopenic Purpura (TTP).
- Idiopathic Thrombocytopenic Purpura (ITP).
Hemophilia
• Definition: disease associated with prolonged bleeding due to the deficiency in clotting factor.• Hemophilia is a X-linked disease. Abnormal gene on X-chromosome
• Types of Hemophilia:
- Hemophilia A
-- Factor 8 deficiency, x linked disease
- Hemophilia B
-- Factor 9 deficiency, x-linked disease
- Hemophilia C
-- Factor 11 deficiency, autosomal genetic disorder
• Symptoms of hemophilia:
- Bruising
- Bleeds easily
- Bleeding into a joint
- Bleeding into the muscles
- Bleeding from injury or bleeding in the brain
- Other sources of bleeding (eg. Stool & urine)
Von Willebrand disease
• The most common hereditary coagulation abnormality• Can also be acquired as a result of other medical conditions
• Due to the deficiency of von Willebrand factor (vWF)
• Von Willebrand factor - mediates binding of glycoprotein Ib to collagen
• This binding mediate activation of platelets and formation of primary hemostasis
• Defect in this factor, resulting glycoprotein IB does not bind to collagen.
• Thus unable to activate platelets, primary hemostasis does not occur
Deficiency of Vitamin K
• Role of Vitamin K in blood coagulation:- Important in maturation of clotting factor.
- modification of certain proteins required for blood coagulation
• If the clotting factor does not mature, it is useless in the hemostasis process.
• Factor which causes the deficiency of vitamin K - Disturbed intestinal uptake.
- By therapeutic or accidental intake of vitamin k-antagonists or very rarely.
- By nutritional vitamin k deficiency
• Some of the possible symptoms of vitamin K deficiency:
- Risk of massive uncontrolled bleeding
Defective Capillary Contractility
• This condition is known as Purpura• Having spontaneous Haemorrhages beneath the skin, mucous membrane.
THROMBOTIC THROMBOCYTOPENIC PURPURA (TTP)
• A blood disorder that causes blood clots to form in small blood vessels around the body, and leads to a low platelet count.• The two main types of TTP are inherited and acquired.
• In inherited TTP, the ADAMTS13 gene is faulty and doesn't prompt the body to make a normal ADAMTS13 enzyme. As a result, enzyme activity is lacking or changed.
• Acquired TTP is the more common type of the disorder. The
ADAMTS13 gene isn't faulty. Instead, the body makes antibodies (proteins) that block the activity of the ADAMTS13 enzyme.
• A lack of activity in the ADAMTS13 enzyme causes TTP.
• The ADAMTS13 gene controls the enzyme, which is involved in blood clotting.
• The enzyme breaks up a large protein called von Willebrand factor that clumps together with platelets to form blood clots.
IDIOPATHIC THROMBOCYTOPENIC PURPURA (ITP)
• Also known as immune thrombocytopenic purpura, is classified as an autoimmune disease.
• The term "idiopathic" indicates that the disease is of an unknown cause or origin: in other words, modern medicine has not yet figured out what it is.
• And the word "purpura" comes from a description of the bruisecolored skin of someone afflicted with the disease: the purple color caused by blood that leaked under the skin.
IDIOPATHIC THROMBOCYTOPENIC PURPURA (ITP)
• Idiopathic thrombocytopenic purpura is a bleeding disorder in which the immune system destroys platelets• Persons with the disease have too few platelets in the blood
• The two types of ITP are acute (temporary or short-term) and chronic (long-lasting).
- Acute ITP generally lasts less than 6 months.
- Chronic ITP lasts 6 months or longer and mostly affects adults.
• Symptoms:
- Abnormally heavy menstruation.
- Bleeding into the skin causes a characteristic skin rash that looks like pinpoint red spots.
- Easy bruising.
- Nosebleed or bleeding in the mouth.

No comments:
Post a Comment