Introduction
• The main function of red blood cell• Transfer of O2 from lungs to tissue
• Transfer of CO2 from tissue to lungs
• To accomplish this function red cells has haemoglobin (Hb)
• Each red cell has 640 million molecules of Hb
The red, oxygen carrying pigment in the RBCs is Haemoglobin. • Structure: 2 parts : heme + globin
• Globin: four chains.
• Heme: porphyrin ring with central iron. Iron is the site of attachment with O2.
• There are 4 heme groups each attached to on globin chain. So one Hb molecule can carry up to 4 O2 molecules.
• According to sequence of amino acids in the primary structure of each chain, there are four types of chains; α, β, γ and δ.
Synthesis
• Synthesis begins in proerythroblast- 65% at erythroblast stage
- 35% at reticulocyte stage
• Haem & globin produced at two different sites in the cells
- Haem in mitochondria
- Globin in polyribosomes

Normal Values
• The hemoglobin level is expressed as the amount of hemoglobin in grams (gm) per deciliter (dL) of whole blood, a deciliter being 100 milliliters.• The normal ranges for hemoglobin depend on the age and, beginning in adolescence, the gender of the person. The normal ranges are:
• Newborns: 17 to 22 gm/dL
• One (1) week of age: 15 to 20 gm/dL
• One (1) month of age: 11 to 15 gm/dL
• Children: 11 to 13 gm/dL
• Adult males: 14 to 18 gm/dL
• Adult women: 12 to 16 gm/dL
• Men after middle age: 12.4 to 14.9 gm/dL
• Women after middle age: 11.7 to 13.8 gm/dL
Types of Hb
• Hb A or HbA1: is the normal Hb in adults represents about 97% of total Hb. it is composed of 2 a and 2 ß chains.• HbA2: minor adult Hb, comprised 3% of normal adult Hb. Composed of 2 a and 2 δ chains
• HbF(fetal Hb): is the main Hb during fetal life and about 60% of normal Hb at birth then disappear gradually. It is composed of 2a and 2 γ chains.
• Hb F has greater affinity for O2 than HbA so ensure O2 transfer from maternal circulation to fetus RBCs through placenta.
Functions of Haemoglobin
• Oxygen delivery to the tissues and CO2 to lungs• Act as excellent acid-base buffer.
• Helps in vasodilation by binding to NO.
• Reaction of Hb & oxygen
- One Hb can bind to four O2 molecules
- Less than .01 sec required for oxygenation
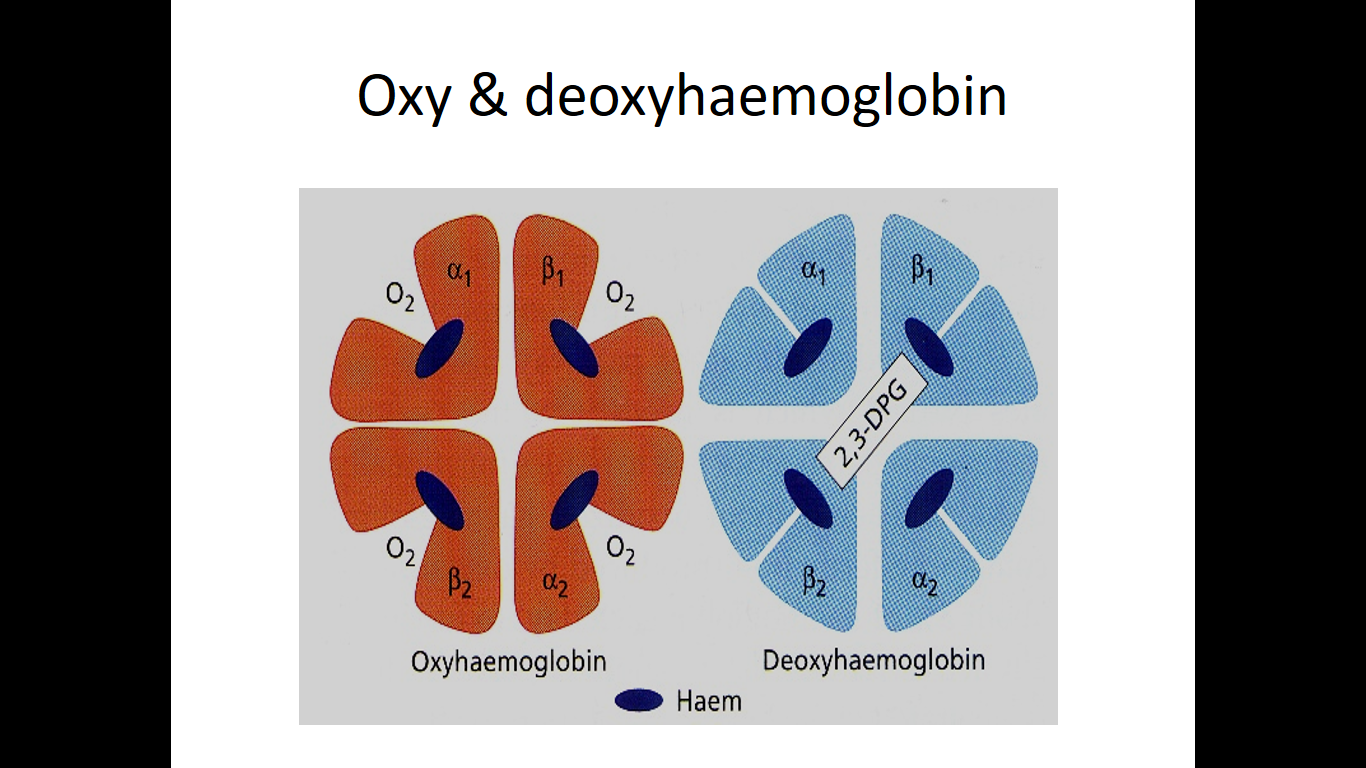
- Oxygenation
- b chain move closer when oxygenated
- When oxygenated 2,3-DPG is pushed out
- b chains are pulled apart when O2 is unloaded, permitting entry of 2,3-DPG resulting in lower affinity of O2
Oxygen-haemoglobin dissociation curve
• O2 carrying capacity of Hb at different Po2• Sigmoid shape
- Binding of one molecule facilitate the second molecule binding
- P 50 (partial pressure of O2 at which Hb is half saturated with O2) 26.6mmHg

• The normal position of curve depends on
- Concentration of 2,3-DPG
- H+ ion concentration (pH)
- CO2 in red blood cells
- Structure of Hb
• Right shift (easy oxygen delivery)
- High 2,3-DPG
- High H+
- High CO2
- HbS
• Left shift (give up oxygen less readily)
- Low 2,3-DPG
- HbF
Different Conjugated Hb
• Carbamino Hb- Reaction of CO2 with Hb• Carboxy Hb or Carbon Monoxy Hb- CO reacts with Hb. Affinity of Hb for CO is 250 times more than O2 which may affect O2 carrying capacity.
• Methaemoglobin- Exposure to various drugs or oxidising agent such ferrous to Ferric.
(Oxidation)
Mutations in hemoglobin (hemoglobinopathies:
1- Sickle cell anemia (Hb S disease):It is a genetic disorder of blood caused by mutation in ß-globin chain resulting in the formation of Hb S. The mutation occurs in 6th position of ß-chain where glutamic acid is replaced by valine (non polar). RBCs assume sickle-shaped leading to fragility of their walls and high rate of hemolysis.

Such sickled cells frequently block flow of blood in narrow capillaries and block blood supply to tissue (tissue anoxia) causing pain and cell death.
Note: The lifetime of erythrocyte in sickle cell is less than 20 days, compared to 120 days for normal RBCs.
Patients may be :
- Heterozygotes (Hb AS): mutation occurs only in one ß-globin chain. These patients have sickle cell trait with no clinical symptoms and can have normal life span.
Or: Homozygotes (Hb SS): mutation occurs in both ß-globin chain with apparent anemia and its
symptoms
2- Hb C disease: Like HbS, Hb C is a mutant Hb in which glutamic acid in 6th position of ß-chain is replaced by lysine. RBCs will be large oblong and hexagonal.
The heterozygous form (HbAC) is asymptomatic.
The homozygous form (Hb CC) causes anemia, tissue anoxia and severe pain.
3- Thalassemia: A group of genetic diseases in which a defect occur in the rate of synthesis of one or more of Hb chains, but the chains are structurally normal. This due to defect or absence of one or more of genes responsible for synthesis of a or ß chains leading to premature death of RBCs.
Types:
ß -thalassemia: When synthesis of ß chains is decreased or absent.There are two copies of the gene responsible for synthesis of ß chains. Individuals with ß globin gene defects have either :
-ß -thalassemia minor (ß -thalassemia trait) : when the synthesis of only one ß -globin gene is defective or absent. Those individuals make some ß chains and usually not need specific treatment.
-ß -thalassemia major ( Cooley anemia): if both genes are defective. Babies will be severely anemic during the first or second year of life and so require regular blood transfusion. Bone marrow replacement is more safe treatment.
-a-thalassemia: in which synthesis of a globin chain is defective or absent. There are four copies of gene responsible for synthesis of a globin chains so patients may have:
-
i - Silent carrier of a-thalassemia with no symptoms: if one gene is defective
ii- a-thalassemia trait: if two genes are defective.
iii- Hb H disease: if 3 a globin genes are defective, with mild to moderate anemia. The produced Hb will be ß4 which is called HB H. Oxygen delivery to tissues will be blocked because Hb H (ß4 ) has high affinity to O2 and not deliver it to tissues.
iv- Hydrops fetalis: when all 4 a globin genes are defective. It causes fetal death because a globin chains are required for synthesis of Hb F.
No comments:
Post a Comment